


医疗器械“钱”途光明,你跟上合规的步伐了吗?

 10230
10230今天我们不吝笔墨,把各大站点关于医疗保健设备的法规、产品认证要求一一罗列,建议大家收藏后仔细阅读哦~
阅读提要:
1)美国:
美国FDA认证
美国FCC认证
2)加拿大
CMDCAS认证
IC认证
3)欧盟CE认证
4)澳洲TGA认证

美国FDA认证

FDA是美国食品和药品管理局(Food and Drug Administration)的缩写。
FDA的官方网站为www.fda.gov
FDA对所有进口到美国或者在美国市场流通的食品和药品,医疗器械和具有医疗功能的保健器械进行管制和规范。
FDA将医疗器械按照危险等级划分为Class I, Class II和Class III, 危险等级逐次提高。FDA对医疗保健设备的分级被规范在FDA官网下的CDRH数据库中,每一种医疗器械都对应一个product code,并且给出申请的指导(guide),提示申请者应该符合哪些程序以及提交哪些资料到FDA进行审核。
FDA的认证程序为:
申请者在FDA CDRH中确定待申请的医疗保健设备所属的product code,并且确定对应的医疗器械分级。
根据FDA CDRH的guide准备申请文件,根据不同分级可能包括a) 电气安全性测试报告; b) 产品设计原理和说明; c) 被FDA批准的其他同类医疗器械的类同性说明; d) 如果涉及到软件分析和控制,需要提交软件设计原理; e) 大部分产品需要提交临床测试报告和分析报告;
提交所有资料到FDA申请批准,在收到完整资料之后FDA会给予申请者一个最终的批准号(K号),该号码也是最终批准之后在FDA网站上列名的注册号,但是在获得FDA最终批准以前不得使用。
FDA进行文件审核并对提交文件中的疑惑问题进行质疑,申请者需要在规定的时间内(90个日历日)对FDA提出的问题进行解答或者补充资料。(FDA的文件审核周期一般为30个日历日每轮)
FDA对文件审核通过之后,会安排进行工厂检查,按照FDA的GMP程序(Good Manufacture Practices),由FDA自行安排。
在GMP工厂检查通过之后15个日历日内,FDA会正式批准该医疗器械申请并且把注册信息显示在FDA官网上供公众查询,产品可以合法在美国市场进行销售和流通。
风险提示
没有获得FDA批准的医疗器械一般不能通过美国海关的报关审核,并且销售/使用方需要对因此产生的医疗事故负担全部民事和刑事责任。
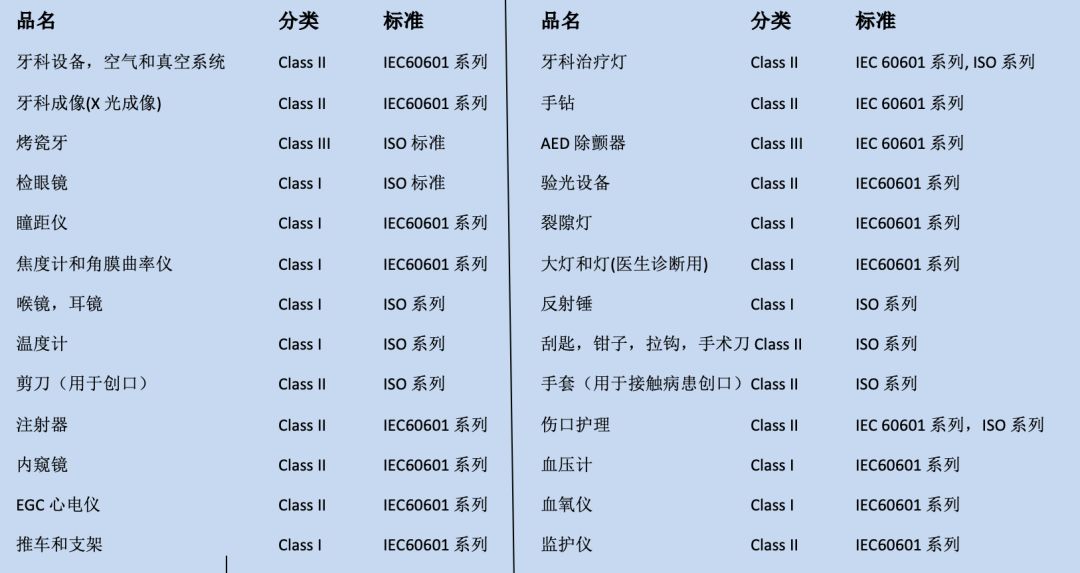
FDA对涉及产品的分类和适用标准:
美国FCC认证

FCC是美国联邦通讯委员会(Federal Communications Commission)的缩写。
FCC的官方网站为www.fcc.gov
FCC负责全美境内的频率分配和合理合法使用,所有使用无线电频率的医疗器械产品都需要符合FCC中47 Code of Federal Regulation Part 18的要求。
FCC的认证程序为:
申请者选择被FCC认可的实验室进行测试并获得通过的报告。(FCC认可的实验室可以在FCC官网进行查询)
准备除了报告以外的相关技术文件,包括说明书,电路图,关键元器件清单,标签等。
完成报告和文件准备之后做符合性自我申明,并且在产品上标注FCC标签(如文中左上标志)(对于有意辐射体/简单说无线电设备,需要符合FCC Certification Procedure,产品需要在本体上打FCC ID号码二不是FCC Logo)
风险提示
没有获得FCC认证的适用医疗器械一般不能通过美国海关的报关审核,并且销售/使用方需要对因此产生的事故或者不良影响负担全部民事和刑事责任。

CMDCAS认证
加拿大医疗器械注册有加拿大卫生局(Health Canada)负责,采取政府注册和第三方质量体系审查的方式。这里所指的第三方,是指被加拿大医疗器械认证认可机构(CMDCAS, Canadian Medical Devices Conformity)认可的第三方机构。
加拿大卫生局的官方网站是www.hc-sc.gc.ca
类似美国,加拿大也对医疗器械采取分级制度,分为Class I, Class II, Class III和Class IV, 风险等级逐次提高。
Class I,即一类医疗器械豁免注册,在具有CMDCAS认可的质量体系证书后可以直接销售,二三四类医疗器械需要进行注册。
需要提交的资料和CE的资料类似,不同等级的医疗器械需要提交的资料要求也不相同,可以在Health Canada网站上检索到具体要求。
具体的注册流程如下:
申请者根据Health Canada网站上的guide确定医疗器械分级(有对应列表确定)
对于非Class I产品,准备质量体系证书和技术资料,递交Health Canada进行注册
Health Canada进行文件审核,通过后予以注册并且签发医疗器械许可证
产品可以合法在加拿大销售。
医疗器械许可证发布后,每年11月1日需要向加拿大卫生部进行再确认。如果于当年12月31日尚未进行再确认的,医疗器械许可证(及营业执照相关范围)自动作废。
CMDCAS对涉及产品的分类和适用标准:

风险提示
没有进行CMDCAS注册的医疗器械一般不能通过的报关审核,并且销售/使用方需要对因此产生的医疗事故负担全部民事和刑事责任。
IC认证
加拿大IC和美国FCC的测试报告技术等同,对于获得FCC认证或测试报告的产品,如果为有意发射体(无线电设备),需要提交资料到加拿大工业局(Industry of Canada, IC)进行换证,如果为无意发射体(常规电子产品),需要在原报告中增加加拿大标准(IC),技术内容等同FCC标准。获得IC证书(或者报告)后产品即可合法在加拿大销售。
参考的官网网站为www.ic.gc.ca

欧盟CE认证

CE认证是European Conformity的法文缩写,是产品进入欧盟境内的必要认证,也包括医疗器械和保健产品欧盟相关技术规定可以在www.cenelec.org上查询。
CE是欧盟于1993年正式设立的产品合格评定程序,由一系列指导文件,指令性文件和标准构成整个程序。
按照产品的风险程度不同,CE制定了guide to the implementation of directives, 规定了module A到module H的8种准入程序。自A到H风险等级逐次升高。
Module A为我们最常见的DoC程序,从法律角度无需第三方介入,厂家可以自行根据符合性证据(比如自己出具的检测报告)进行自我符合性声明。
Module B ~ Module H分别为EC Type Examination到Full Quality Assurance, 都不允许厂家自我声明产品的符合性,需要进行工厂检查,引入欧盟提名机构(Notified Body)进行文件审核,工厂审核等才能完成符合性程序。
基于选定的认证程序(Module种类),欧盟针对每个类别或者特性的产品规范了具体的符合性要求,这个规范称为欧盟指令(EU Directive),指令中规定了欧盟和在欧盟生产/销售产品的责任机构的责任和义务,并且列出了产品需要符合的一般性技术要求范围(比如安全性,电磁兼容,有毒有害物质管控,废弃电子电器回收,能效,噪音,排放等等),适用的产品需要符合指令规定的所有相关技术要求。
每一个技术要求范围,在指令中一般都不给于具体的测试要求或者标准,这些要求和标准会被规范在指令的下一级文件(标准和执行决议)(Standard and implementation)中,这些被指令涵盖的标准和执行文件每年会由欧盟发布对应指令的年刊(Official Journey, OJ)给出详细列表。
执行中,申请者需要根据产品特性选定适用的指令,并找到OJ中列出的相关标准进行检测证明符合性(对于不在OJ中列出但是必要的安全性标准,欧盟Notified body会给出建议),在符合通过后根据选定的Module自我宣告或者递交欧盟相关Notified body审核,并且在通过审核后根据Module中的规定在产品本体上贴上CE标签,或者CE标签加欧盟Notified Body编号以完成CE符合性程序的所有工作。
欧盟对医疗器械进行分级管理制度,按照危险等级不同按照93/42/EEC医疗器械指令分为General Class I, Special Class I; Class IIA, Class IIB, Class III类产品, 级别越高危险程度越高。医疗器械指令Article 19明确规定了各种医疗器械的分级要求。
医疗保健产品的认证程序为:
申请者按照医疗器械指令确定待申请的医疗保健设备所属的医疗器械分级。
根据不同分级准备文件资料,可能包括a) 测试报告可能包括安全性,电磁兼容性,无线电特性,化学有毒有害物质管控,废弃电子电器产品回收测试分析,能效评估; b) 产品设计原理和说明; c) 被FDA批准的其他同类医疗器械的类同性说明; d) 如果涉及到软件分析和控制,需要提交软件设计原理; e) 大部分产品需要提交临床测试报告和分析报告;
准备有效的ISO13485工厂质量体系证书
将文件和工厂质量体系证书提交欧盟提名机构(Notified Body)进行审核
审核过程中Notified Body会就提交的资料进行审核和提出质疑,申请者需要及时进行回复解释或者补充提交资料
在Notified Body审核通过以后,会颁发CE证书,申请产品本体需要标注该Notified body在欧盟的注册编号以标示具体哪个Notified Body进行了审核和批准(比如Intertek在医疗领域的Notified Body号码是英国0473或者瑞典0413)
在获得CE证书后产品可以合法向欧洲销售和流通。
风险提示
没有获得Notified Body批准的医疗器械一般不能通过欧盟海关的报关审核,并且销售/使用方需要对因此产生的医疗事故负担全部民事和刑事责任,后果严重!
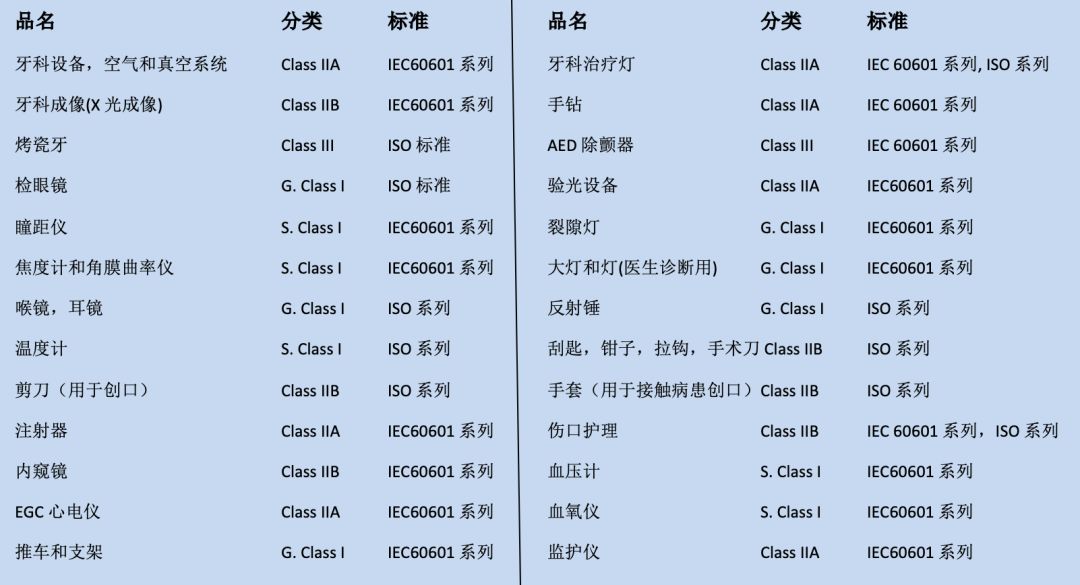
CE对涉及产品的分类和适用标准:

澳洲TGA认证

澳洲医疗用品管理局(Therapeutic Goods Administration)是负责对进口到澳洲的医疗器械,药品和保健产品进行注册和管制的机构, 官方网站为www.tga.gov.au 。
澳洲对医疗器械准入和市场监管通过TGA下的两个部门分管,其中ODA (office of devices authorization)负责产品销售前的批准(对海外产品也就是准入),OPR(office of product review)负责产品投放市场之后的监督管理。
澳洲对医疗器械的分级制度和欧盟相同,在表述上分为Class I, Class Is, Cass Im, Class IIA, Class IIB和Class III, 危险程度逐次提高。其中Class Is是指带有消毒功能(sterile)的一类医疗设备(比如消毒柜),Class Im是指带有测量读数功能(measurement)的一类医疗设备(比如水银体温表),欧盟把这两类都简化为特殊一类医疗器械。
对一类医疗器械澳洲指管制产品符合性,对除了一类和三类以外的其他医疗器械管制产品符合性和工厂质量管理体系(ISO13485),对于三类医疗器械管制产品符合性,工厂质量管理体系和设计控制(Design/type control)
澳洲TGA的注册程序为:
申请者按照上述TGA规定确定医疗产品等级
申请者提供包括被TGA认可的符合性测试报告或者欧盟Notified Body出具的CE证书及其所有对应技术资料和报告,以及澳洲符合性声明到TGA进行审核
TGA进行审核,如果TGA提出质疑可能需要补充材料,如果TGA通过审核,申请者上传资料到ARTG (Australian Register of Therapeutic Goods)完成注册
在ARTG上显示注册成功之后,信息可以被公众查询到并且产品可以合法的投入市场
备注: 此处的申请者是指投放市场或者分销的澳洲法人,可以是位于澳洲的制造商,或者是位于澳洲的分销商,但是不是澳洲以外的制造商。
澳洲TGA的具体定义,要求和程序可以在ARGMD(Australia Registration Guide of Medical Devices)上查询到,包括准入要求、市场监督和管理要求等细节。
风险提示
没有获得TGA批准的医疗器械一般不能通过澳洲海关的报关审核,并且销售/使用方需要对因此产生的医疗事故负担全部民事和刑事责任。
GA对涉及产品的分类和适用标准:

备注: 对于非医疗器械产品,澳洲有独立的RCM认证程序进行管制,根据产品定义来选择是RCM还是TGA程序。
注:由于各个国家、地区的认证方式不一,且随时将由不同机构进行修改,本指南所列的认证方式仅供参考,最新及详细的步骤请参见各国官方网站。
-END-




今天我们不吝笔墨,把各大站点关于医疗保健设备的法规、产品认证要求一一罗列,建议大家收藏后仔细阅读哦~
阅读提要:
1)美国:
美国FDA认证
美国FCC认证
2)加拿大
CMDCAS认证
IC认证
3)欧盟CE认证
4)澳洲TGA认证
美国FDA认证
FDA是美国食品和药品管理局(Food and Drug Administration)的缩写。
FDA的官方网站为www.fda.gov
FDA对所有进口到美国或者在美国市场流通的食品和药品,医疗器械和具有医疗功能的保健器械进行管制和规范。
FDA将医疗器械按照危险等级划分为Class I, Class II和Class III, 危险等级逐次提高。FDA对医疗保健设备的分级被规范在FDA官网下的CDRH数据库中,每一种医疗器械都对应一个product code,并且给出申请的指导(guide),提示申请者应该符合哪些程序以及提交哪些资料到FDA进行审核。
FDA的认证程序为:
申请者在FDA CDRH中确定待申请的医疗保健设备所属的product code,并且确定对应的医疗器械分级。
根据FDA CDRH的guide准备申请文件,根据不同分级可能包括a) 电气安全性测试报告; b) 产品设计原理和说明; c) 被FDA批准的其他同类医疗器械的类同性说明; d) 如果涉及到软件分析和控制,需要提交软件设计原理; e) 大部分产品需要提交临床测试报告和分析报告;
提交所有资料到FDA申请批准,在收到完整资料之后FDA会给予申请者一个最终的批准号(K号),该号码也是最终批准之后在FDA网站上列名的注册号,但是在获得FDA最终批准以前不得使用。
FDA进行文件审核并对提交文件中的疑惑问题进行质疑,申请者需要在规定的时间内(90个日历日)对FDA提出的问题进行解答或者补充资料。(FDA的文件审核周期一般为30个日历日每轮)
FDA对文件审核通过之后,会安排进行工厂检查,按照FDA的GMP程序(Good Manufacture Practices),由FDA自行安排。
在GMP工厂检查通过之后15个日历日内,FDA会正式批准该医疗器械申请并且把注册信息显示在FDA官网上供公众查询,产品可以合法在美国市场进行销售和流通。
风险提示
没有获得FDA批准的医疗器械一般不能通过美国海关的报关审核,并且销售/使用方需要对因此产生的医疗事故负担全部民事和刑事责任。
FDA对涉及产品的分类和适用标准:
美国FCC认证
FCC是美国联邦通讯委员会(Federal Communications Commission)的缩写。
FCC的官方网站为www.fcc.gov
FCC负责全美境内的频率分配和合理合法使用,所有使用无线电频率的医疗器械产品都需要符合FCC中47 Code of Federal Regulation Part 18的要求。
FCC的认证程序为:
申请者选择被FCC认可的实验室进行测试并获得通过的报告。(FCC认可的实验室可以在FCC官网进行查询)
准备除了报告以外的相关技术文件,包括说明书,电路图,关键元器件清单,标签等。
完成报告和文件准备之后做符合性自我申明,并且在产品上标注FCC标签(如文中左上标志)(对于有意辐射体/简单说无线电设备,需要符合FCC Certification Procedure,产品需要在本体上打FCC ID号码二不是FCC Logo)
风险提示
没有获得FCC认证的适用医疗器械一般不能通过美国海关的报关审核,并且销售/使用方需要对因此产生的事故或者不良影响负担全部民事和刑事责任。
CMDCAS认证
加拿大医疗器械注册有加拿大卫生局(Health Canada)负责,采取政府注册和第三方质量体系审查的方式。这里所指的第三方,是指被加拿大医疗器械认证认可机构(CMDCAS, Canadian Medical Devices Conformity)认可的第三方机构。
加拿大卫生局的官方网站是www.hc-sc.gc.ca
类似美国,加拿大也对医疗器械采取分级制度,分为Class I, Class II, Class III和Class IV, 风险等级逐次提高。
Class I,即一类医疗器械豁免注册,在具有CMDCAS认可的质量体系证书后可以直接销售,二三四类医疗器械需要进行注册。
需要提交的资料和CE的资料类似,不同等级的医疗器械需要提交的资料要求也不相同,可以在Health Canada网站上检索到具体要求。
具体的注册流程如下:
申请者根据Health Canada网站上的guide确定医疗器械分级(有对应列表确定)
对于非Class I产品,准备质量体系证书和技术资料,递交Health Canada进行注册
Health Canada进行文件审核,通过后予以注册并且签发医疗器械许可证
产品可以合法在加拿大销售。
医疗器械许可证发布后,每年11月1日需要向加拿大卫生部进行再确认。如果于当年12月31日尚未进行再确认的,医疗器械许可证(及营业执照相关范围)自动作废。
CMDCAS对涉及产品的分类和适用标准:
风险提示
没有进行CMDCAS注册的医疗器械一般不能通过的报关审核,并且销售/使用方需要对因此产生的医疗事故负担全部民事和刑事责任。
IC认证
加拿大IC和美国FCC的测试报告技术等同,对于获得FCC认证或测试报告的产品,如果为有意发射体(无线电设备),需要提交资料到加拿大工业局(Industry of Canada, IC)进行换证,如果为无意发射体(常规电子产品),需要在原报告中增加加拿大标准(IC),技术内容等同FCC标准。获得IC证书(或者报告)后产品即可合法在加拿大销售。
参考的官网网站为www.ic.gc.ca
欧盟CE认证
CE认证是European Conformity的法文缩写,是产品进入欧盟境内的必要认证,也包括医疗器械和保健产品欧盟相关技术规定可以在www.cenelec.org上查询。
CE是欧盟于1993年正式设立的产品合格评定程序,由一系列指导文件,指令性文件和标准构成整个程序。
按照产品的风险程度不同,CE制定了guide to the implementation of directives, 规定了module A到module H的8种准入程序。自A到H风险等级逐次升高。
Module A为我们最常见的DoC程序,从法律角度无需第三方介入,厂家可以自行根据符合性证据(比如自己出具的检测报告)进行自我符合性声明。
Module B ~ Module H分别为EC Type Examination到Full Quality Assurance, 都不允许厂家自我声明产品的符合性,需要进行工厂检查,引入欧盟提名机构(Notified Body)进行文件审核,工厂审核等才能完成符合性程序。
基于选定的认证程序(Module种类),欧盟针对每个类别或者特性的产品规范了具体的符合性要求,这个规范称为欧盟指令(EU Directive),指令中规定了欧盟和在欧盟生产/销售产品的责任机构的责任和义务,并且列出了产品需要符合的一般性技术要求范围(比如安全性,电磁兼容,有毒有害物质管控,废弃电子电器回收,能效,噪音,排放等等),适用的产品需要符合指令规定的所有相关技术要求。
每一个技术要求范围,在指令中一般都不给于具体的测试要求或者标准,这些要求和标准会被规范在指令的下一级文件(标准和执行决议)(Standard and implementation)中,这些被指令涵盖的标准和执行文件每年会由欧盟发布对应指令的年刊(Official Journey, OJ)给出详细列表。
执行中,申请者需要根据产品特性选定适用的指令,并找到OJ中列出的相关标准进行检测证明符合性(对于不在OJ中列出但是必要的安全性标准,欧盟Notified body会给出建议),在符合通过后根据选定的Module自我宣告或者递交欧盟相关Notified body审核,并且在通过审核后根据Module中的规定在产品本体上贴上CE标签,或者CE标签加欧盟Notified Body编号以完成CE符合性程序的所有工作。
欧盟对医疗器械进行分级管理制度,按照危险等级不同按照93/42/EEC医疗器械指令分为General Class I, Special Class I; Class IIA, Class IIB, Class III类产品, 级别越高危险程度越高。医疗器械指令Article 19明确规定了各种医疗器械的分级要求。
医疗保健产品的认证程序为:
申请者按照医疗器械指令确定待申请的医疗保健设备所属的医疗器械分级。
根据不同分级准备文件资料,可能包括a) 测试报告可能包括安全性,电磁兼容性,无线电特性,化学有毒有害物质管控,废弃电子电器产品回收测试分析,能效评估; b) 产品设计原理和说明; c) 被FDA批准的其他同类医疗器械的类同性说明; d) 如果涉及到软件分析和控制,需要提交软件设计原理; e) 大部分产品需要提交临床测试报告和分析报告;
准备有效的ISO13485工厂质量体系证书
将文件和工厂质量体系证书提交欧盟提名机构(Notified Body)进行审核
审核过程中Notified Body会就提交的资料进行审核和提出质疑,申请者需要及时进行回复解释或者补充提交资料
在Notified Body审核通过以后,会颁发CE证书,申请产品本体需要标注该Notified body在欧盟的注册编号以标示具体哪个Notified Body进行了审核和批准(比如Intertek在医疗领域的Notified Body号码是英国0473或者瑞典0413)
在获得CE证书后产品可以合法向欧洲销售和流通。
风险提示
没有获得Notified Body批准的医疗器械一般不能通过欧盟海关的报关审核,并且销售/使用方需要对因此产生的医疗事故负担全部民事和刑事责任,后果严重!
CE对涉及产品的分类和适用标准:
澳洲TGA认证
澳洲医疗用品管理局(Therapeutic Goods Administration)是负责对进口到澳洲的医疗器械,药品和保健产品进行注册和管制的机构, 官方网站为www.tga.gov.au 。
澳洲对医疗器械准入和市场监管通过TGA下的两个部门分管,其中ODA (office of devices authorization)负责产品销售前的批准(对海外产品也就是准入),OPR(office of product review)负责产品投放市场之后的监督管理。
澳洲对医疗器械的分级制度和欧盟相同,在表述上分为Class I, Class Is, Cass Im, Class IIA, Class IIB和Class III, 危险程度逐次提高。其中Class Is是指带有消毒功能(sterile)的一类医疗设备(比如消毒柜),Class Im是指带有测量读数功能(measurement)的一类医疗设备(比如水银体温表),欧盟把这两类都简化为特殊一类医疗器械。
对一类医疗器械澳洲指管制产品符合性,对除了一类和三类以外的其他医疗器械管制产品符合性和工厂质量管理体系(ISO13485),对于三类医疗器械管制产品符合性,工厂质量管理体系和设计控制(Design/type control)
澳洲TGA的注册程序为:
申请者按照上述TGA规定确定医疗产品等级
申请者提供包括被TGA认可的符合性测试报告或者欧盟Notified Body出具的CE证书及其所有对应技术资料和报告,以及澳洲符合性声明到TGA进行审核
TGA进行审核,如果TGA提出质疑可能需要补充材料,如果TGA通过审核,申请者上传资料到ARTG (Australian Register of Therapeutic Goods)完成注册
在ARTG上显示注册成功之后,信息可以被公众查询到并且产品可以合法的投入市场
备注: 此处的申请者是指投放市场或者分销的澳洲法人,可以是位于澳洲的制造商,或者是位于澳洲的分销商,但是不是澳洲以外的制造商。
澳洲TGA的具体定义,要求和程序可以在ARGMD(Australia Registration Guide of Medical Devices)上查询到,包括准入要求、市场监督和管理要求等细节。
风险提示
没有获得TGA批准的医疗器械一般不能通过澳洲海关的报关审核,并且销售/使用方需要对因此产生的医疗事故负担全部民事和刑事责任。
GA对涉及产品的分类和适用标准:
备注: 对于非医疗器械产品,澳洲有独立的RCM认证程序进行管制,根据产品定义来选择是RCM还是TGA程序。
注:由于各个国家、地区的认证方式不一,且随时将由不同机构进行修改,本指南所列的认证方式仅供参考,最新及详细的步骤请参见各国官方网站。
-END-


 热门活动
热门活动 
 福建
福建 01-08 周四
01-08 周四
 热门报告
热门报告 










