




全球速卖通出口欧盟商品资质管控--医疗器械出口法规须知


全球速卖通发布关于加强出口欧盟商品资质管控公告,公告如下:


商品合规一直是速卖通关注重点,为了应对当下全球消费者的需求增长,保障商家安全公平的交易环境、提升消费者体验以及满足各国合规和安全要求,速卖通要求商家发布的商品符合目的市场的相关法律及规定。任何销往欧洲地区的商品需要严格符合欧盟相关的商品合规及商品安全要求。
平台拟进一步加强售往欧盟的婴幼儿玩具、电子电器、小家电、装饰灯具、医疗器械、化妆品、珠宝饰品、运动户外、安全防护等品类的商品管控。商家需确保自己提供的商品符合销售国当地的相关法律法规。
具体类目的合规要求,平台将结合各国合规及法律安全要求,对于不满足目的国合规要求的商品(包括但不限于缺失CE证书,EMC/LVD检测报告,ROHS检测报告及商品外包装标签图(包含产品信息、生产企业信息、欧代责任人信息以及资质Logo及必要警示语)等,速卖通可能对相关商品屏蔽区域化市场,对于恶意规避的商品将执行下架扣分等处罚。
今天让我们来了解下医疗器械出口法规须知。
医疗器械在美国上市主要需根据美国联邦法规法典 第21册 - 食品和药品中的要求进行管控,该法规规定了医疗器械的定义,分类,管控方式。其他还可能涉及到的法规有:
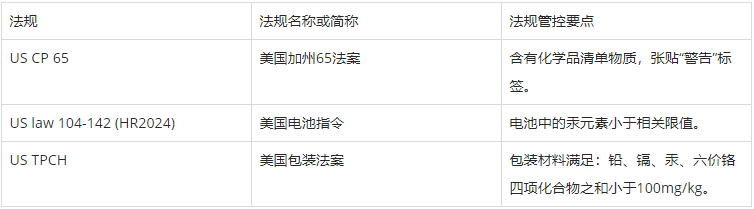
US CP 65法案是美国加州基于保护加州公民饮用水安全制定的相关法案。法案规定了相关企业必须针对法案化学品清单物质履行“警告”义务。
US law 104-142 (HR2024)指令规定了美国境内电池产品相关管控要求。指令规定了相关电池产品相关有害物质限制要求。
US TPCH法案规定了美国境内包装材料的相关管控要求。法案规定了包装材料相关有害物质的限制要求。
医疗器械在欧盟上市主要需符合以下法规:
医疗器械法规Medical Device Regulation EU 2017/745
预期取代医疗器械指令Medical Devices Directive COUNCIL DIRECTIVE 93/42/EEC和有源植入医疗器械指令AIMD Council Directive 90/385/EEC
体外诊断医疗器械法规IVDR EU2017/746
预期取代体外诊断医疗器械指令 IVDD,Council Directive 98/79/EEC
法规规定了医疗器械的定义,分类规则,及合规流程。
其他还可能涉及到的法规有:
2011/65/EU 指令规定了欧盟境内供应和销售的电子电气产品相关有害物质限制要求。
(EC) No.1907/2006法规规定了欧盟境内生产、制造和销售所有物质、混合物和物品的相关化学品限制要求。相关物品需要满足法规Article 33关于SVHC的要求以及Article 67关于附录XVII的要求。
(EU) No.2019/1021法规规定了欧盟境内所有物质、混合物和物品中相关持久性有机污染物的限制要求。
2012/19/EU指令规定了欧盟境内废弃电子电气产品相关管控要求。指令涉及到回收标识和回收率等相关规定。
2006/66/EC指令规定了欧盟境内相关电池产品的相关管控要求。指令涉及到相关限用物质限制要求和标识要求。
94/62/EC指令规定了欧盟境内相关包装材料的相关管控要求。指令规定了包装材料相关有害物质的限制要求。
美国的医疗器械是指符合以下条件的仪器、装置、工具、机具、器具、插入管、体外试剂或其它相关物品,包括组件、零件或附件等;意图使用于人类疾病或其它身体状况的诊断;或用于疾病之治愈、减缓、治疗者;意图影响人类身体的功能或结构,但不经由人类身体或身体上的化学反应来达成其首要目的,同时也不依赖新陈代谢来达成其主要目的。
欧盟的医疗器械新法规对医疗器械的定义修改如下:是指单独或者组合使用于人体的任何仪器、设备、器具、软件、植入物、试剂、材料或其他物品;其作用于人体体表或体内的主要效用不是通过药理学、免疫学或者代谢的手段获得,但可能有这些手段参与并起一定的辅助作用;旨在达到下列一个或多个目的:疾病的诊断、预防、监护、治疗或缓解;损伤或者残疾的诊断、监护、治疗、缓解或修补;解剖学和生理或病理过程或状态的探查、替代或调节;来自器官、血液和组织捐献的人体标本体外检验数据的提供;专门用于对医疗器械清洗、消毒或灭菌和对妊娠的控制和支持的器械应被认为是医疗器械。
1. 美国:
美国联邦法规法典第 21 册-食品和药品中对医疗器械按照风险等级做了分类,低风险的器械归为I类,中等风险的器械归为II类,高风险的器械归为III类。针对具体器械进行了规则编号,且每一种器械指定了器械代码,并指定了产品需要符合的相关标准。
a. 低风险I类
I类为“普通管理”产品,是指危险性小或基本无危险性的产品,它的设计一般比Ⅱ类产品、Ⅲ类产品简单。FDA 认为绝大多数的 I 类产品通过一般控制足以保证其安全性和有效性。I 类产品要求符合一般控制,例如医用手套、压舌扳、手动手术器械等。管控要求为一般管控,即在FDA进行注册列名。
b. 中风险Ⅱ类
Ⅱ类是指那些用一般控制不足以控制其安全性和有效性,必须通过现有的其他方式,即特殊控制,来保证其安全性和有效性的产品。例如体温计,心电图仪、超声诊断仪、输血输液器具、呼吸器等。在Ⅱ类产品市场准入前,一般需申请市场准入前通告 510(k)。
c. 高风险Ⅲ类
Ⅲ类是指那些仅用一般控制和特殊控制还不足以确保其安全性和有效性的产品。这类产品具有较大危险性或危害性,它一般用来支持人体生命,防止人体健康受损,具有致病、致残的潜在的、不合理的风险。例如人工心脏瓣膜、心脏起搏器、人工晶体、人工血管等。FDA对这类产品实行上市前批准(PMA)制度。
另外,关于化学方面,还可能涉及:
2. 欧盟:
欧盟医疗器械法规将医疗器械分成Ⅰ,Ⅱa,Ⅱb 和Ⅲ类等四个类别:
Ⅰ 类为非侵入式器械,除用于储存血液,与Ⅱa以上类型医疗器械联用,改变液体成分,与真皮层接触的器械外。
Ⅱa 类包括诊断设备、体液储存、输入器械,以及短暂使用(持续时间小于 1h)并有侵害性的外科器械。
Ⅱb 类为短期使用(持续时间 lh~30d)并有侵害性的外科用器械、避孕用具和放射性器械。
Ⅲ类器械为与中枢神经系统、心脏接触的器械、在体内降解的器械、植入体内的器械和药物释放器械,以及长期使用(持续时间大于30d)并有侵害性的外科器械。
a. 低风险I类
医疗器械指令/法规明确规定了I类医疗器械的分类规则,需要符合的基本安全和性能要求及评估流程。I类医疗器械包括I类测量器械(Im),I类灭菌器械(Is),和I类其他器械;I类其他器械可以出具自我符合性声明来满足CE要求;I类测量器械(Im),I类灭菌器械(Is)则需要公告机构(Notified body)签发CE认证证书。
b. 中风险II类
医疗器械指令/法规明确规定了II类医疗器械的分类规则,需要符合的基本安全和性能要求及管控要求。须向公告机构提出上市申请,由公告机构负责审查(Ⅱa 类产品的产品设计由生产企业负责,公告机构主要检查其质量体系;Ⅱb 类产品由公告机构审查质量体系、抽检样品,同时生产企业应提交产品设计文件),通过审查后,签发CE认证证书,产品贴 CE 标志,方可进入欧盟各成员国市场。
c. 高风险III类
医疗器械指令/法规明确规定了Ⅲ类医疗器械的分类规则,需要符合的基本安全和性能要求及管控要求。Ⅲ类产品由公告机构审查,要检查质量体系、抽检样品,并审查产品设计文件,特别是审查产品风险分析报告。通过审查后,签发CE认证证书,产品贴 CE 标志,方可进入欧盟各成员国市场。
另外,关于化学方面,还可能涉及:

为了帮助各位商家更好的合规,平台特甄选了合规服务商入驻服务市场,为商家提供服务。如果商家有合规需求,可以通过服务市场下单咨询。
很荣幸,“雅玛森跨境”成为了全球速卖通甄选的合规服务商之一,我们将以专业的服务帮助平台卖家完成税务登记,获得平台及当地法律要求的税号证书,并根据你在平台的销售额和收入完成月度、季度和年度纳税申报;提供例如EPR注册等合规性的服务,确保您的商品符合世界各地的规定和法规要求。








")























