





一文读懂系列丨欧洲CE注册要求&流程解读

“市场概况”
据MedTec Europe的数据,2022年欧洲医疗器械市场的总规模达到了1350亿欧元,占全球市场的27%,位居全球第二,仅次于美国。
欧洲法规变革:从2021年5月开始,欧洲的医疗器械生产商必须符合2017/745号欧盟医疗器械法规,而非先前的93/42/EEC医疗器械指令,以获得CE标志的授权。在此背景下,产品必须依据MDR进行分类。不过,请注意,部分分类规则并未改变,您可以参考以下MDD的分类路径。
欧洲市场准入要求:如果您是制造商,计划将医疗设备销售到欧盟,您需确保产品符合欧盟委员会的相关欧洲指令,如医疗器械指令(MDD): AIMDD90/385/EEC、MDD93/42/EEC、IVDMDD98/79/EC。为证明产品满足CE指令的基本准则,必须在产品上附加CE标志,并须通过CE认证标记流程。这一过程将根据医疗器械的类型和您选择的合格评定路径来决定。产品的具体特性,例如预期用途、侵入性和效果等,将决定其分类和对患者的风险程度。
医疗器械分类:欧洲将医疗器械分为四大类别:I类、IIa类、IIb类和III类,其中III类风险最高。由于新监管体系的更严格要求,许多设备的分类已有所调整。例如,一些之前归于IIa或IIb类的产品现在被归入III类。如果您的医疗设备不属于I类,您需要向认证机构提交证据,证明产品符合相应CE指令的基本准则。
一类医疗器械的CE标记流程
一类医疗器械在风险等级上属于最低级别。制造商在为这类设备申请CE标记时,有三种不同的途径可以选择。决策时,需要考虑如下几个因素:首先,该医疗器械是否是无菌的,比如个人防护装备;其次,是否具有测量功能,例如听诊器;最后,如果既不是无菌也不具备测量功能,如矫正眼镜。值得注意的是,如果您的一类产品既不是无菌的,也不是测量设备,您可以通过自我认证的方式,通过书面声明确认其符合MDD的相关要求。若为无菌或测量类医疗器械,则需经认证机构评估。
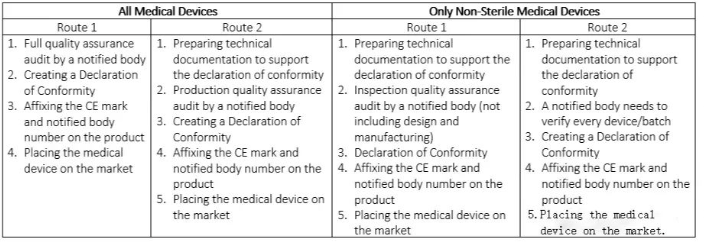
表1:I类医疗器械的CE认证标志路线
关于IIa类医疗器械的CE认证
IIa类医疗器械,例如手术手套、助听器和诊断超声机,一般属于中低风险级别,通常供患者短期使用,不得超过30天。如果您是制造这类医疗设备的厂商,您必须提交相应的证明,符合认证机构的评估标准,才能让产品上市。有四种不同的方法来申请CE标志,这些方法可以根据产品是否无菌分为两组。
表2.IIa类医疗器械的CE标志路线
IIb类医疗器械的CE认证流程
IIb类医疗器械包括诸如长期矫正隐形眼镜、手术激光器、除颤器等产品,这些属于中高风险设备,患者可能会使用超过30天。如果您的产品属于IIb类别,您将需要通过指定机构评估您的技术文件,以确保它们符合医疗器械指令的规定。
您的产品类型将再次决定特定CE标志路径的选择。
壁当啷响,我总爱跟你谈及宇宙,温柔,橘子汽水味儿的风和蓝色的落日。
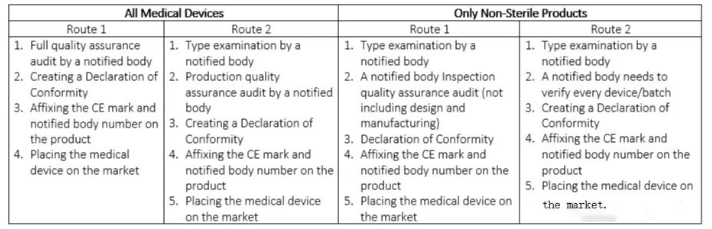
表3.IIb类医疗器械的CE标志路线
第三类医疗器械的CE认证详解
第三类医疗器械涵盖风险最高的医疗设备,例如心血管导管、动脉瘤夹、髋关节植入物、人工心脏瓣膜等,需在其整个生命周期中持续受到监控。有专门机构负责对这些产品进行严格审查和监督。
与II类医疗器械类似,第三类医疗器械的合格评估可能涉及对技术文件的全面审查和质量体系或产品的检查,特别关注设备的设计和生产方面。
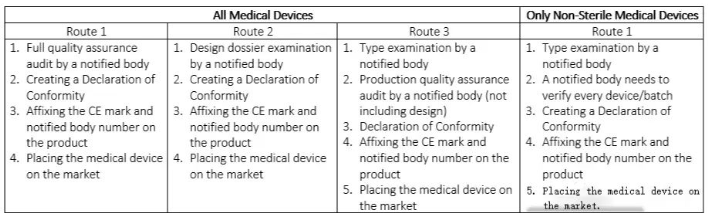
表4三类医疗器械的CE标志路线






")





















